
Projects
Our main aim is to develop and apply computer-aided methods to model, simulate and predict how biomolecules interact. The focus is on the interactions of proteins. The methods make use of three-dimensional macromolecular structures and combine approaches based on physicochemical principles with those of chemo- and bio-informatics.
Some of our projects are described on this page.
CompIF: Efficient computation of intermolecular forces for biomolecular simulations
Molecular simulations enable the structure, dynamics, and interactions of biomolecules to be explored in detail. Trajectories (or movies) of the evolution …
Read more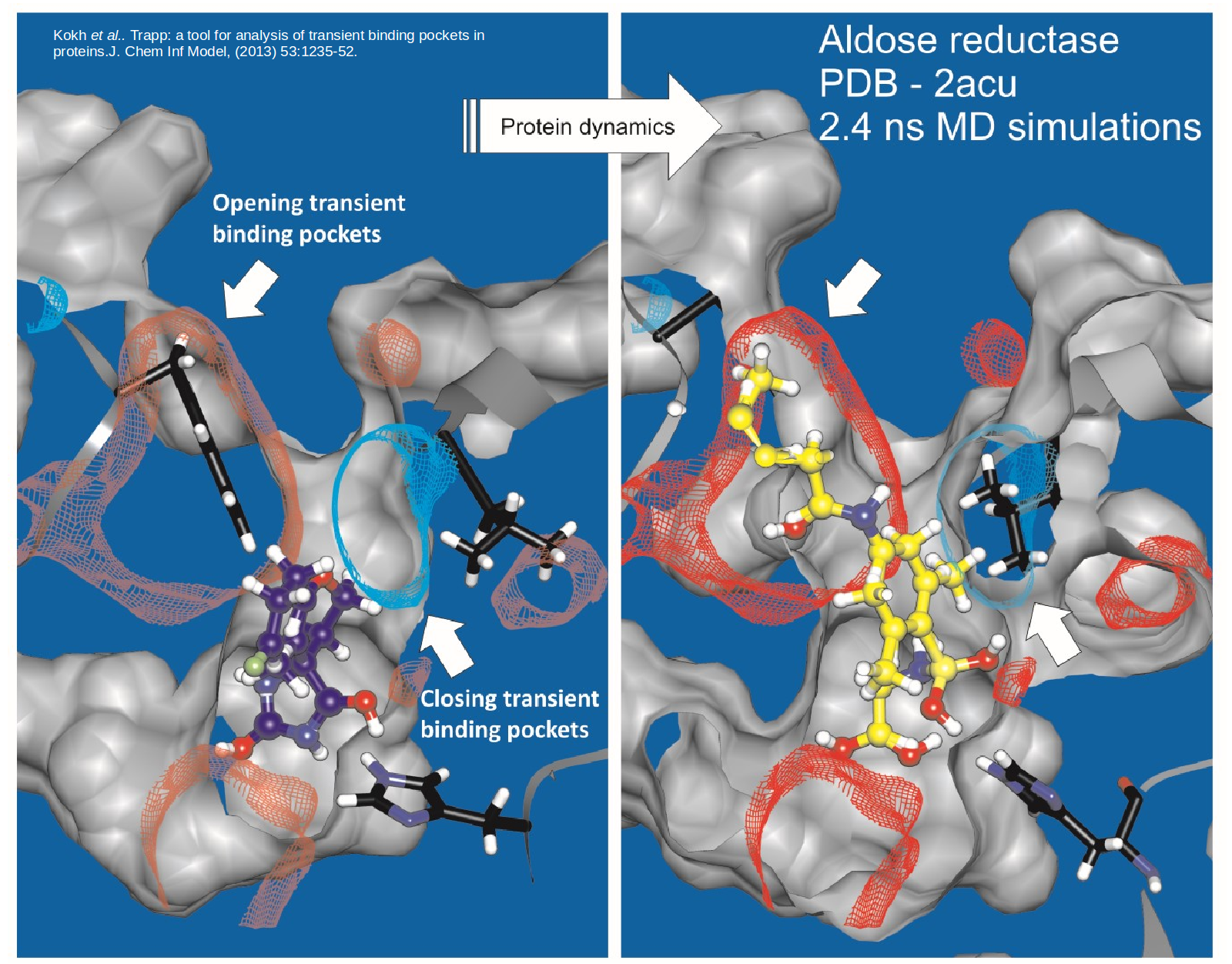
Structure-based drug discovery
Proteins are dynamic and constantly changing their shape. This flexibility not only presents a challenge to to structure-based drug design approaches …
Read more
Macromolecular interactions and diffusional association
We are developing methods to predict protein-protein interactions and how proteins bind to surfaces and membranes. These methods mostly rely on …
Read more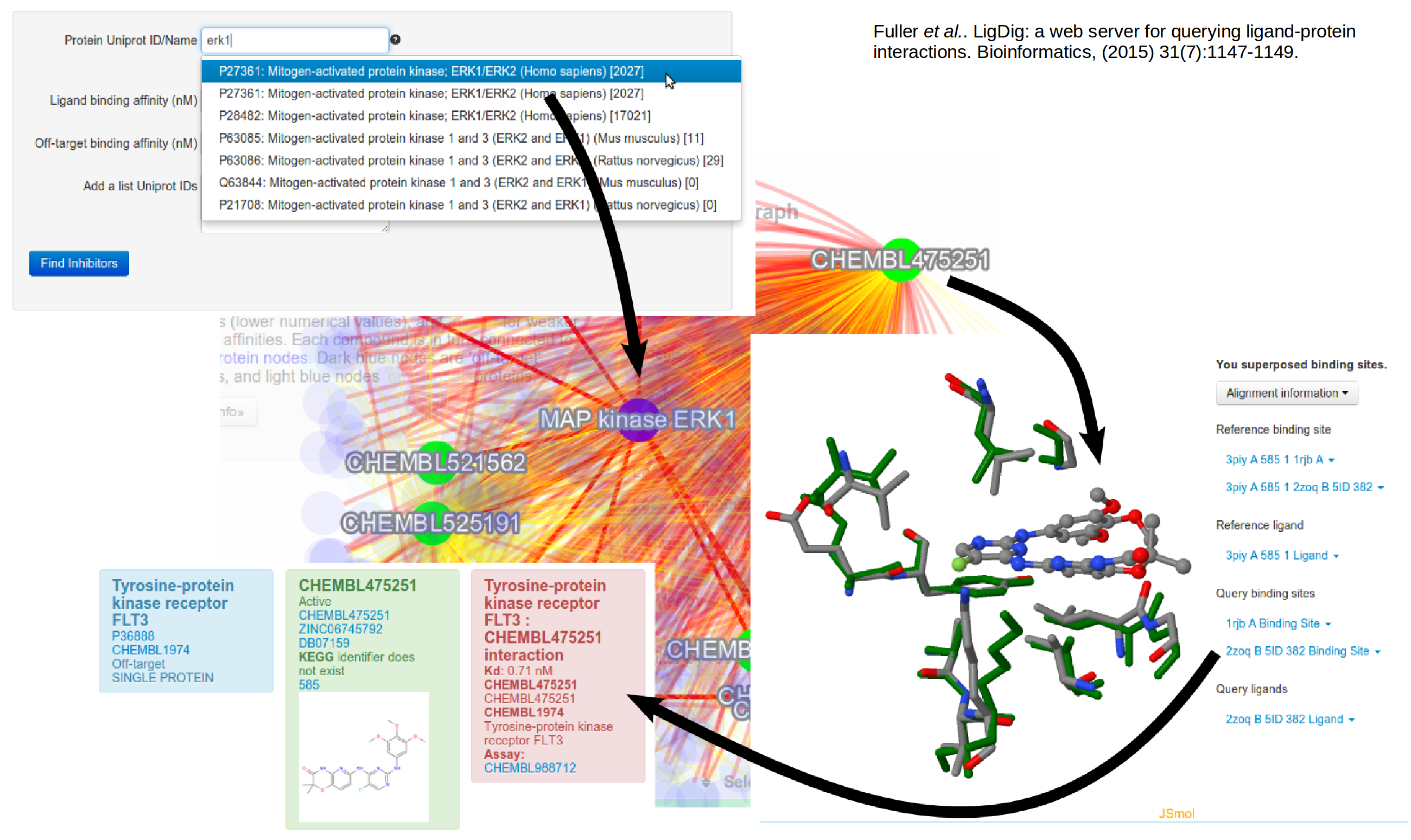
Protein structures in systems biology
We are working on developing approaches to bridge between protein structures and biochemical networks, from the molecular to the cellular level. …
Read moreGallery

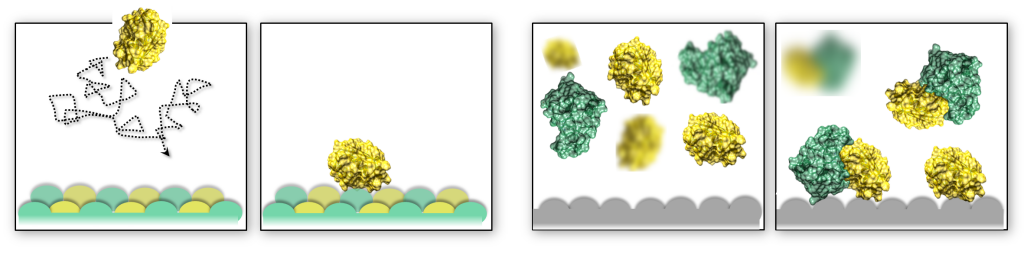
The shape of macromolecular crowders affects protein diffusion 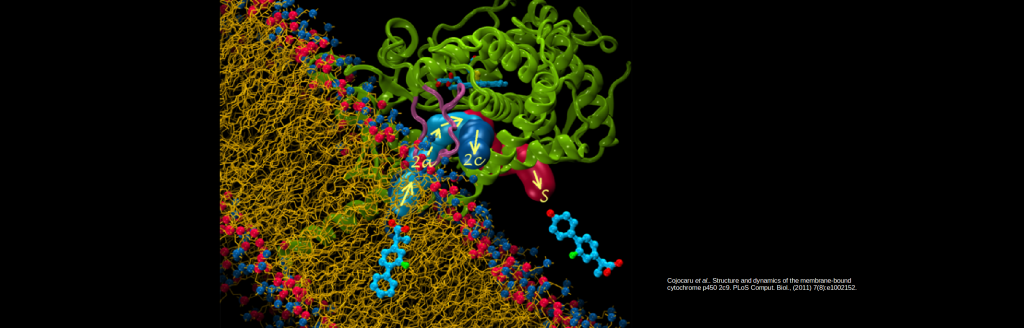
Simulations identify routes in and out of cytochrome P450 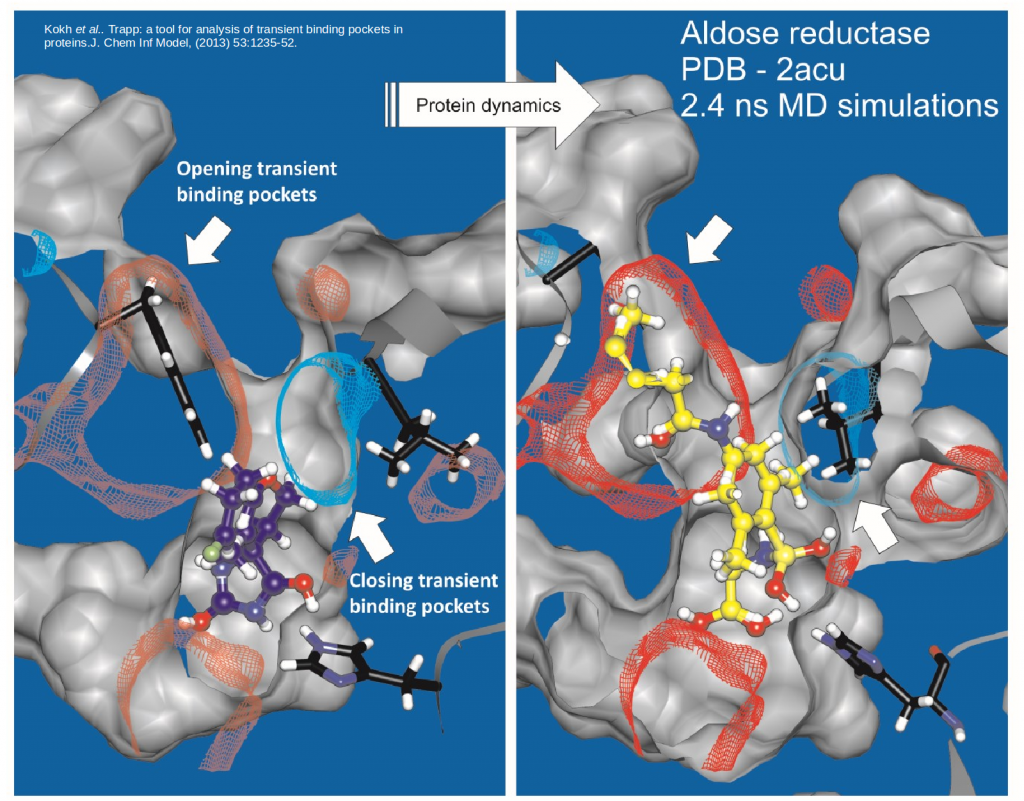
TRAPP helps to find transient pockets in proteins 
Simulations show how sickle cell hemoglobin starts to oligomerize 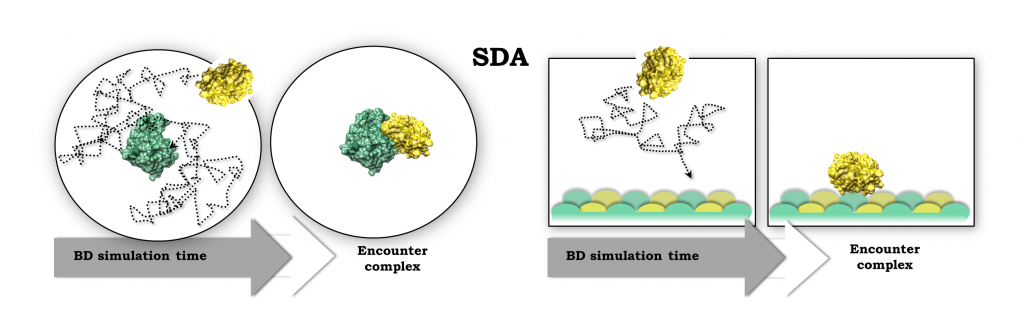
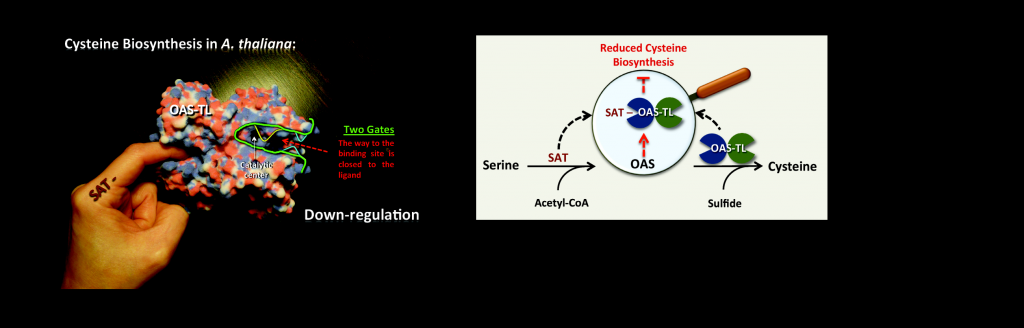
Allosterically gated enzyme dynamics regulate cysteine biosynthesis 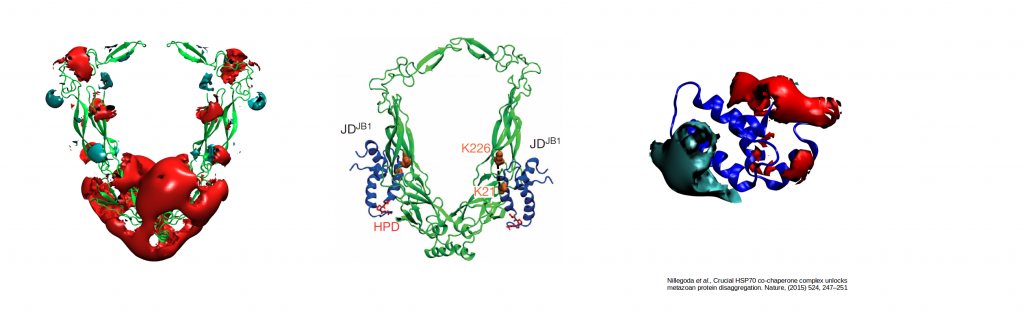
Electrostatically-guided Brownian dynamics docking of two co-chaperone J-proteins 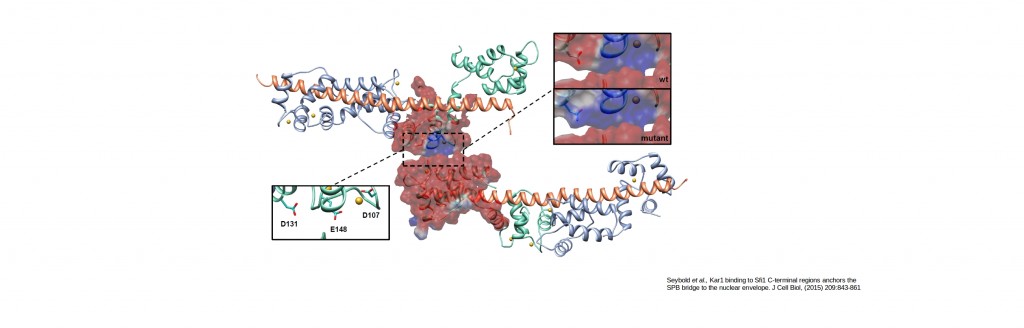
Free energy calculations for proteins stabilizing the spindle pole body bridge