Structure-based drug discovery
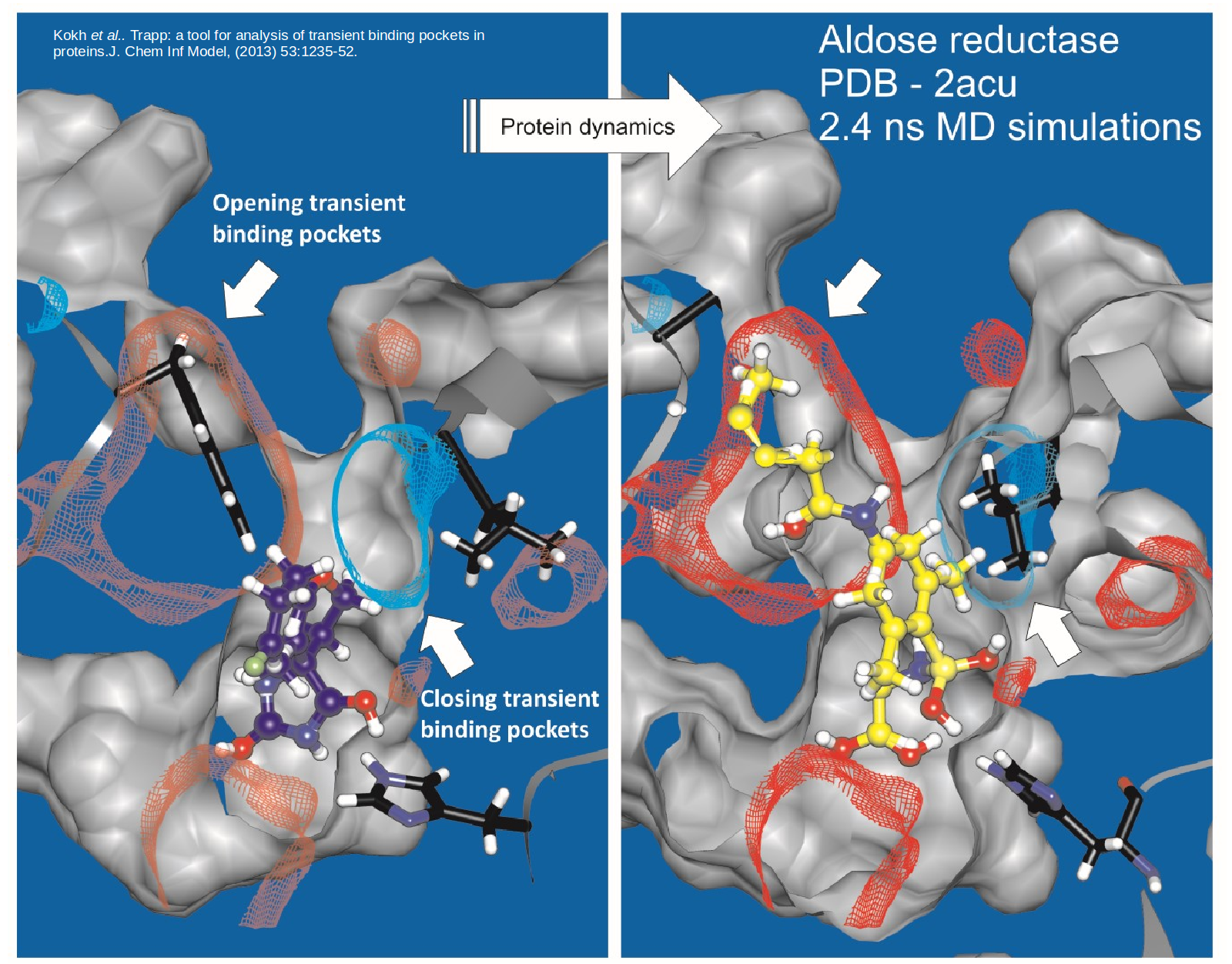
Proteins are dynamic and constantly changing their shape. This flexibility not only presents a challenge to structure-based drug design approaches but also opportunities for the design of specific compounds with suitable kinetic properties. We are developing methods to model and simulate protein and ligand dynamics in order to identify transient binding pockets in proteins and to compute the kinetics of drug binding.
TRAPP is a Tool for Analysis of Transient Binding Pockets in Proteins that is available for web server or standalone use. It automates the tracking, analysis, and visualization of binding pocket variations along a protein motion trajectory or within an ensemble of protein structures (Kokh et al, 2013 ; Stank et al., 2017). To efficiently generate protein conformations that reveal transient pockets, TRAPP incorportates two methods that we developed based on the Rotationally-induced perturbation (RIP) simulation method: L-RIP and RIPlig (Kokh et al., 2016). TRAPP computes a druggability score for individual binding site conformations, thus enabling the identification of potentially druggable transient conformations or cryptic pockets (Yuan et al., 2020).
The importance of binding kinetics for drug discovery has recently become appreciated and was the motivation for the K4DD IMI project aimed at developing methods to measure and predict drug-target binding kinetics as well as understand quantitative structure-kinetics relationships (QSKRs). We are developing methods to simulate protein-drug binding and unbinding processes and to predict the corresponding on-rates and off-rates. Among these is tauRAMD for computing relative residence times (Kokh et al., 2018). For recent applications, see: Berger et al. 2021 and Nunes-Alves et al. 2021, as well as the KBbox toolbox.
We apply computational methods to challenging drug discovery problems. We currently contribute to EuroNeurotrophin – a European training network for the discovery of neurotrophins small molecule mimetics as potential therapeutic agents for neurodegeneration and neuroinflammation. We work on the structure-based design of peptidomimetics against striated muscle disorders in the Informatics4Life joint initiative. We participated in the NMTRypI New Medicines for Trypanosomal Infections EU project to identify new candidate drugs against neglected parasitic diseases such as leishmaniasis.