SDA7 – Brownian Dynamics Simulations of Proteins: New features
Researchers from the Molecular and Cellular Modeling (MCM) group at HITS are developing the software “Simulation of Diffusional Association” (SDA) 7.
SDA7 was initially developed to carry out Brownian dynamics (BD) simulations of the diffusional association in a continuum aqueous solvent of two solute molecules, e.g. proteins, or of a solute molecule to an inorganic surface. SDA7 can also used to simulate the diffusion of multiple proteins, in dilute or concentrated solutions, e.g. to study the effect of macromolecular crowding. Two new features have recently been introduced:
- BD–MD Coupling for Drug-Protein Association: A new multiscale pipeline combines the computational efficiency of BD with the accuracy of molecular dynamics (MD). BD handles long-range drug diffusion until an encounter complex is formed near the binding site; MD then captures the short-range atomic interactions needed for forming a fully bound complex. This approach significantly cuts computational cost while yielding association rate constants (kon) that have been validated against experimental data for a range of protein-ligand complexes of varying size and flexibility [1,2].
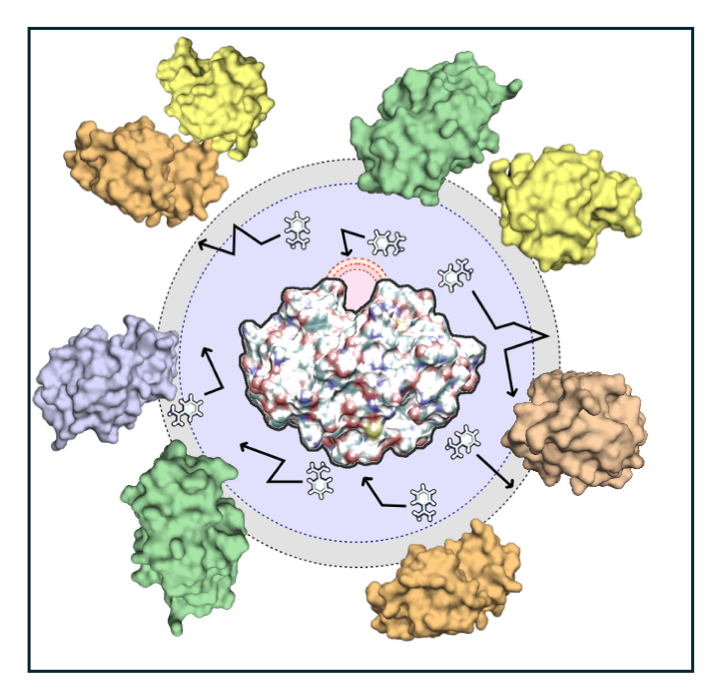
- Multi-Resolution Crowding Model: Inside living cells, molecules diffuse through a dense environment packed with other macromolecules — a phenomenon called macromolecular crowding. SDA7 now includes an adaptive multi-scale model that retains full atomic detail for crowders near the associating molecules, while using a coarse-grained representation for distant ones. Combined with Markov State modelling, this model has been used to investigate the competition between opposing effects on the rate at which a drug binds its target protein: crowders can hinder binding by slowing diffusion, or enhance it through channeling and caging effects — with the net outcome depending on the physicochemical properties of the specific system [3].
The SDA7 software package is available at https://mcm.h-its.org/sda7 and part of the functionality is implemented in the webserver version, webSDA.
References:
[1] Muñiz-Chicharro A, Ganotra GK, Wade RC (2025). A Multiscale Simulation Approach to Compute Protein–Ligand Association Rate Constants by Combining Brownian Dynamics and Molecular Dynamics, J. Chem. Inf. Model. 65(20):11215-11231.
[2] Beccaria R, Muñiz-Chicharro A, Wade RC (2026). How does macromolecular crowding affect protein-Ligand association? A multiscale Brownian dynamics simulation approach. ChemRxiv
DOI: https://doi.org/10.26434/chemrxiv.15004053/v1
[3] Muñiz-Chicharro A, Chernova E, Ganotra GK, Wade RC (2026). SDAMD: a multiscale workflow to compute protein-ligand binding association rate constants using Brownian dynamics and molecular dynamics simulations. Methods Mol. Biol. Accepted.
[4] Muñiz‐Chicharro A, Votapka LW, Amaro RE, Wade RC (2023). Brownian dynamics simulations of biomolecular diffusional association processes, WIREs Comput Mol Sci 13(3),e1649
About HITS
HITS, the Heidelberg Institute for Theoretical Studies, was established in 2010 by physicist and SAP co-founder Klaus Tschira (1940-2015) and the Klaus Tschira Foundation as a private, non-profit research institute. HITS conducts basic research in the natural, mathematical, and computer sciences. Major research directions include complex simulations across scales, making sense of data, and enabling science via computational research. Application areas range from molecular biology to astrophysics. An essential characteristic of the Institute is interdisciplinarity, implemented in numerous cross-group and cross-disciplinary projects. The base funding of HITS is provided by the Klaus Tschira Foundation.