Project 2: Excited states potential energy surfaces and spin flips
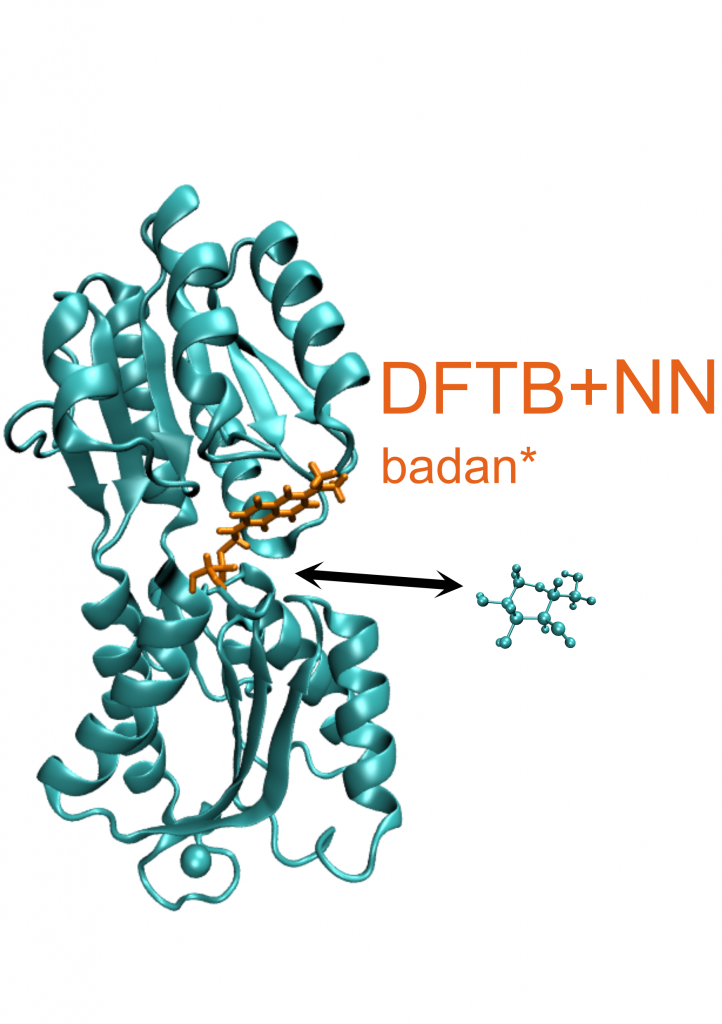
Recently, machine learning models were successfully applied to predict the ground state potential energy surface (PES) of organic molecules and inorganic solids [1,2]. However, excited states PES are significantly more challenging to describe using machine learning models, limiting the application to very small molecules so far [3,4]. A combination of delta ML trained on highly accurate but expensive quantum chemistry calculation and significantly faster DFTB calculations was not tried yet for this challenge. We plan to use active learning approaches to sample the conformational space, and the bias corrected DFTB calculations will then be used to compute trajectories at the semi-classical surface-hopping level to sample the excited-state behavior of the molecules. A particular challenge is the calculations of fluorescence spectra of chromophors in biological molecules, like the badan molecule, which is attached to the glucose binding protein in order to measure phyisoligical glucose concentrations. We will derive ML models and DFTB/ML hybrids to study biological fluorescence pheonomena.
References:
1] Behler, Parrinello, (2007) Generalized neural-network representation of high-dimensional potential-energy surfaces, PRL 98, 146401
[2] Friederich et al., (2020) Machine-learned potentials for next-generation matter simulations, Nature Materials Reviews, 20, pages 750–761 (202)
[3] Westermeyr et al., (2019) Machine learning enables long time scale molecular photodynamics simulations, Chem. Sci. 10, 8100-8107
[4] Westermeyr et al., (2020) Machine learning and excited-state molecular dynamics, Machine Learning: Science and Technology
Team

Prof. Dr. Marcus Elstner
Lead Principal Investigator (Karlsruhe Institute of Technology)
Phone: +49 721 60845705
More Information
T.T.-Prof. Dr. Pascal Friederich
Lead Principal Investigator (Karlsruhe Institute of Technology)
Phone: +49 721 60844764
More Information
Prof. Dr. Andreas Dreuw
Lead Principal Investigator (IWR, Heidelberg University)
Phone: +49 6221 5414735
More Information
Christian C. Schmidt
PhD Student (Karlsruhe Institute of Technology)
Phone: +49 721 608 45706
More Information