New tools for computing protein-ligand dissociation rates and exploring dissociation mechanisms
The length of time that a drug molecule spends bound to its protein target – its residence time – is an important determinant of its efficacy, and is inversely related to the rate of dissociation of the drug-target complex. Consequently, there is a need for computational methods to predict dissociation rates to guide drug design and discovery. For most pharmaceutically relevant compounds, the timescales for dissociation from the target far exceed those that are accessible to conventional molecular dynamics simulation methods.
To address this problem, we developed and evaluated an efficient computational workflow that enables the prediction of relative drug-protein residence times and the analysis of dissociation mechanisms in an automated manner. The workflow is based on simulations performed with the Random Acceleration Molecular Dynamics (RAMD) method which, in addition to existent implementations in the NAMD and AMBER software packages, we have now implemented in the freely available GROMACS molecular simulation engine for simulations on CPU or GPU nodes.
Relative dissociation rates are computed with the tauRAMD protocol and dissociation trajectories are analyzed using protein–ligand interaction fingerprints with our new MD-IFP set of tools. The workflow is described in Kokh et al. J. Chem. Phys. 153, 125102 (2020); doi: 10.1063/5.0019088 , recently published in a JCP Special Topic on Classical Molecular Dynamics (MD) Simulations: Codes, Algorithms, Force fields, and Applications.
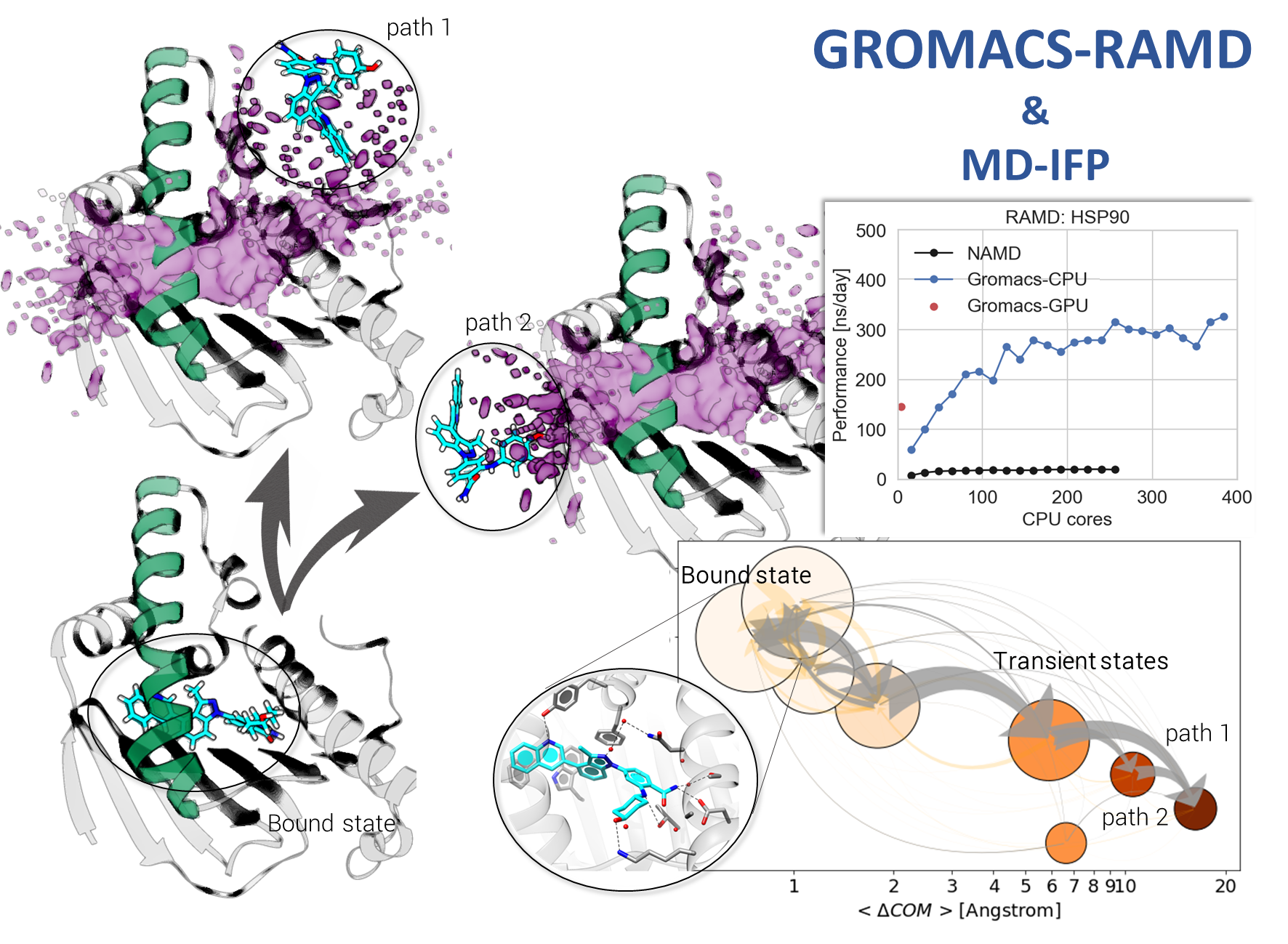
Kokh DB, Doser B, Richter S, Ormersbach F, Cheng X, Wade RC “A workflow for exploring ligand dissociation from a macromolecule: Efficient random acceleration molecular dynamics simulation and interaction fingerprint analysis of ligand trajectories”, J. Chem. Phys. 153, 125102 (2020); doi: 10.1063/5.0019088, recently published in a JCP Special Topic on Classical Molecular Dynamics (MD) Simulations: Codes, Algorithms, Force fields, and Applications.
About HITS
HITS, the Heidelberg Institute for Theoretical Studies, was established in 2010 by physicist and SAP co-founder Klaus Tschira (1940-2015) and the Klaus Tschira Foundation as a private, non-profit research institute. HITS conducts basic research in the natural, mathematical, and computer sciences. Major research directions include complex simulations across scales, making sense of data, and enabling science via computational research. Application areas range from molecular biology to astrophysics. An essential characteristic of the Institute is interdisciplinarity, implemented in numerous cross-group and cross-disciplinary projects. The base funding of HITS is provided by the Klaus Tschira Foundation.