Von Form zu Form: die dynamische Welt der Proteine
Im Juli 2025 entwickelten Forscher*innen des Heidelberger Instituts für Theoretische Studien (HITS) und des Max-Planck-Instituts für Polymerforschung (MPIP) ein Modell, das Proteine mit individuell anpassbarer Flexibilität entwerfen kann. Ein neues, ergänzendes Deep-Learning-Modell – Back Bone Flow (BBFlow) – kann nun die Dynamik von Proteinen vorhersagen und damit Einblicke in ihre Funktionsweise liefern. Die Ergebnisse wurden im Dezember 2025 auf internationalen Fachkonferenzen in San Diego und Kopenhagen vorgestellt.
Anders als die bereits bestehenden Modelle GAFL (Geometric Algebra Flow Matching) und FliPS (Flexibility-Conditioned Protein Structure Design with Flow Matching) kann dieses Flow-basierte Deep-Learning-Modell die dreidimensionale Struktur eines Proteins allein anhand der Geometrie des Rückgrats – der Hauptkette der Atome – erfassen. Bei klassischen Ansätzen werden dafür komplexe Simulationen oder evolutionäre Sequenzdaten benötigt.
„Während BBFlow die Proteindynamik vorhersagt, generiert FliPS neue Proteine mit der gewünschten Dynamik. BBFlow kann dann die besten von FliPS generierten Kandidaten herausfiltern“, sagt Nicolas Wolf, Erstautor der Studie.
Darüber hinaus ist BBFlow leicht zugänglich, intuitiv bedienbar und erfordert keinerlei Programmierkenntnisse. Gleichzeitig arbeitet es bis zu 40-mal schneller als das derzeit führende Modell „AlphaFlow“ – bei vergleichbarer Genauigkeit. Da das Modell keine evolutionären Daten benötigt, eignet es sich besonders für neue oder synthetische Proteine, für die es keine natürlichen Vorbilder gibt.
Schnell, effizient und präzise: eine Bandbreite an Ensembles
Es ist vor allem diese hohe Geschwindigkeit, mit der BBFlow punkten kann. Das Modell lässt sich in wenigen Tagen von Grund auf trainieren, ohne aufwändiges Pre-Training oder evolutionäre Informationen. Tests zeigen, dass es realistische Ensembles sowohl für natürlich vorkommende als auch für neu entworfene (de novo) Proteine erzeugt – auch wenn für diese Proteine keine evolutionären Daten vorliegen.
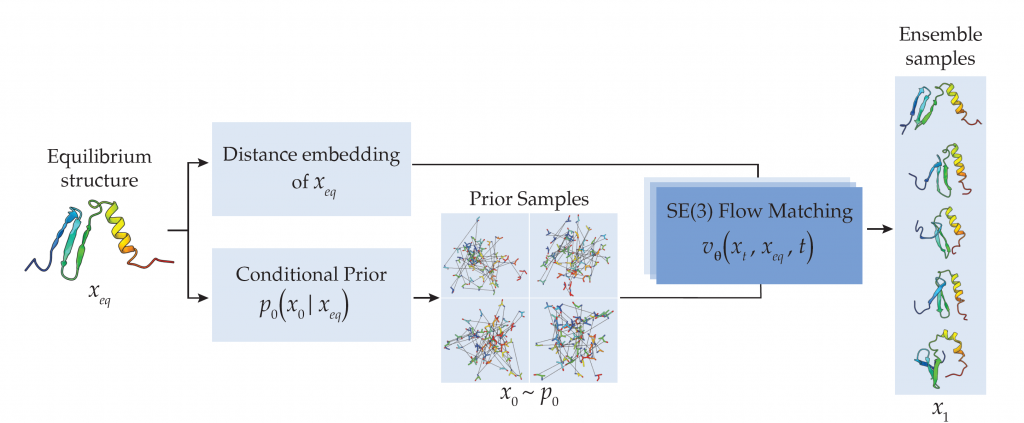
„Statt auf evolutionäre Daten zu setzen, nutzen wir jetzt eine Methode, die Proteinstrukturen direkt erzeugt. Dabei verwenden wir die bekannte Ausgangsstruktur des Proteins, um das Modell zu steuern“, sagt Leif Seute, Doktorand in der Forschungsgruppe Machine Learning and Artificial Intelligence (MLI) und Zweitautor der Studie. Das Ergebnis: deutlich schnellere Vorhersagen bei vergleichbarer Genauigkeit. „Die Methode funktioniert nicht nur für natürliche monomere Proteine, sondern auch für de novo und multimerische Proteine“, fügt Seute hinzu.
Dieser Ansatz könnte die Protein-Forschung und das Design neuer Proteine erheblich beschleunigen – ein wichtiger Schritt für die Entwicklung neuer Medikamente, Therapien und Materialien. Statt nur eine einzige „funktionale Form“ zu liefern, erzeugt BBFlow ein ganzes Ensemble möglicher Strukturen, was die Flexibilität von Proteinen realistischer abbildet.
Ein Schritt hin zu authentischen Proteinmodellen
Indem der Fokus von starren Einzelstrukturen auf dynamische Ensembles verschoben wird, kommen die Forscher*innen der Frage näher, wie Proteine tatsächlich in der Zelle agieren. Auch wenn das aktuelle Modell sich bisher auf das Rückgrat beschränkt und noch nicht alle Atome und Seitenketten einbezieht, zeigt es: die Rückgratgeometrie allein liefert wesentlich mehr Informationen als bislang angenommen. Es eröffnet eine neue Perspektive, in der Wirklichkeitstreue, Geschwindigkeit und Flexibilität beim Protein-Design nicht länger im Widerspruch stehen.
N. Wolf, L. Seute, V. Viliuga, S. Wagner, J. Stühmer, F. Gräter. Learning conformational ensembles of proteins based on backbone geometry. NeurIPS, 2025.
Wissenschaftlicher Kontakt:
Jun.-Prof. Dr. Jan Stühmer
Junior Group Leader
Machine Learning and Artificial Intelligence
Heidelberger Institut für Theoretische Studien (HITS)
https://www.h-its.org/de/people/dr-jan-stuhmer/
Prof. Dr. Frauke Gräter
Director, Abteilungsleiterin “Biomolekulare Mechanik”
Max-Planck-Institut für Polymerforschung (MPIP)
https://www.mpip-mainz.mpg.de/1001466/01_Direktor
Medienkontakt:
Angela Michel
Head of Communications
Heidelberger Institut für Theoretische Studien (HITS)
+49 (0)6221 533 277
angela.michel@h-its.org
Teresa Petry
Presse- und Öffentlichkeitsarbeit
Max-Planck-Institut für Polymerforschung (MPIP)
+49 (0)6131 379-119
pr@mpip-mainz.mpg.de
Über das HITS
Das HITS (Heidelberger Institut für Theoretische Studien) wurde 2010 von dem Physiker und SAP-Mitbegründer Klaus Tschira (1940-2015) und der Klaus Tschira Stiftung als privates, gemeinnütziges Forschungsinstitut gegründet. Es betreibt Grundlagenforschung in den Naturwissenschaften, der Mathematik und der Informatik. Zu den Hauptforschungsrichtungen zählen komplexe Simulationen auf verschiedenen Skalen, Datenwissenschaft und -analyse sowie die Entwicklung rechnergestützter Tools für die Forschung. Die Anwendungsfelder reichen von der Molekularbiologie bis zur Astrophysik. Ein wesentliches Merkmal des Instituts ist die Interdisziplinarität, die in zahlreichen gruppen- und disziplinübergreifenden Projekten umgesetzt wird. Die Grundfinanzierung des HITS wird von der Klaus Tschira Stiftung bereitgestellt.