SS 2022: Lectures and Hands-on sessions in computational molecular biophysics
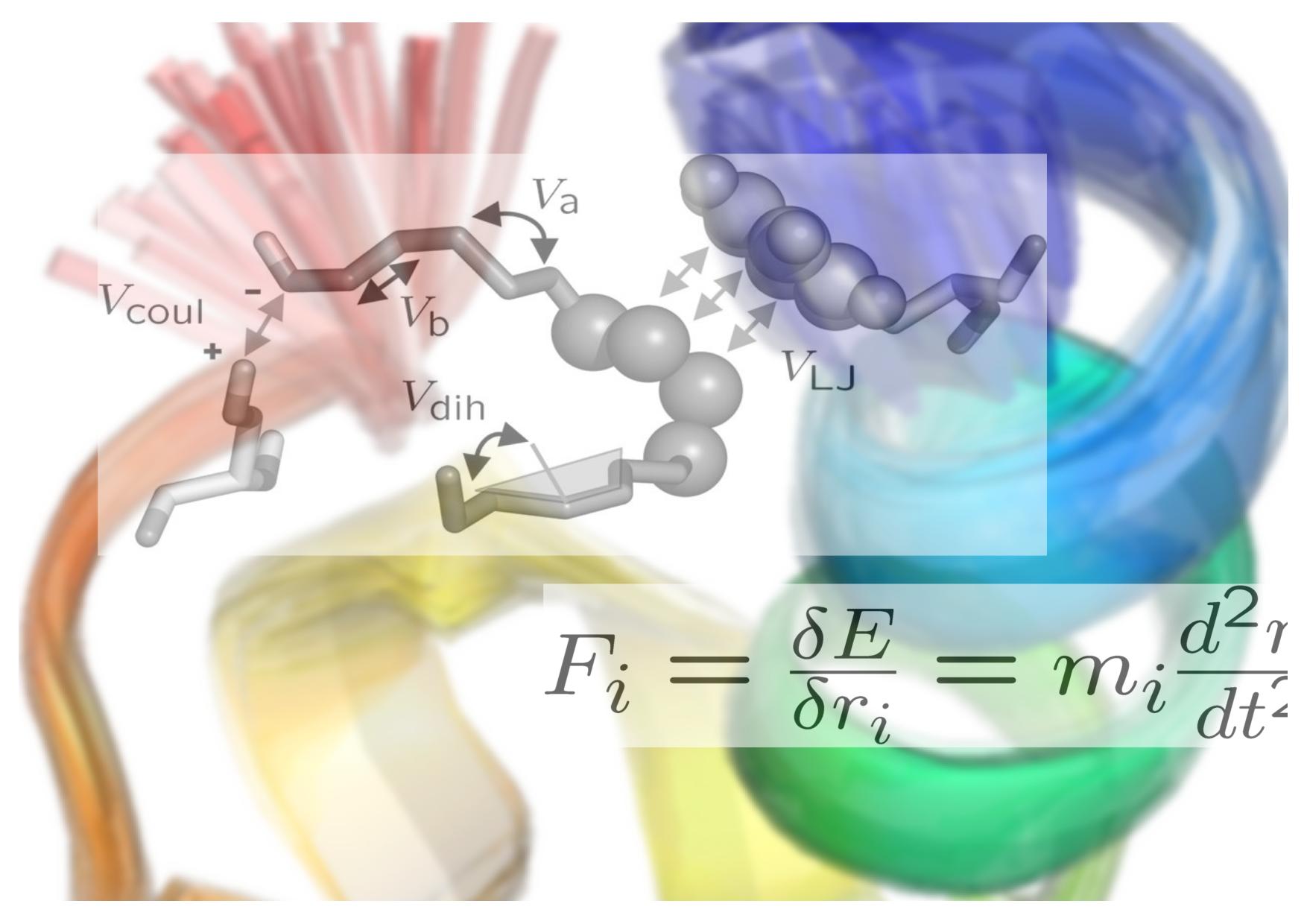
How can a bird sense magnetic fields, how does our ear detect sound waves, how does our bone feel gravitation? It is the physics of individual molecules that ultimately dictate these and many other processes in life.
This course introduces computational methods to study the structure, dynamics and mechanics of biomolecules at different scales. It aims at endowing the students with an active understanding of the principles, the capacity and limitations of different molecular simulation techniques, including Monte Carlo, Molecular Dynamics, Brownian Dynamics, Machine Learning and multi-scale simulations with an emphasis on Molecular Dynamics simulations. The course comprises alternating lectures and hands-on computer tutorials of which the latter are meant to directly demonstrate the principles of running and analyzing computer simulations of biological matter. Lectures will be given by Prof. Frauke Gräter, Prof. Rebecca Wade and Dr. Camilo Aponte-Santamaría.
The lectures are targeted to advanced Bachelor, Master and interested PhD students and will be complemented by hands-on computer sessions in which the students will have the opportunity to run molecular simulations.
Time and place:
The lectures/tutorials will take place once a week, on Thursdays, 9-11 am (2 SWS). The first lecture will be on April 21.
Place of lectures and hands-on computer tutorials: INF 205 / CIP-Pool
6 ECTS (for biologists 4 ECTS) will be awarded.
Only a limited number of students can be accepted. Please register in advance by sending an Email to Frauke.Graeter@h-its.org.
Schedule (tentative): to be announced
Literature
- Recommended: Schlick, “Molecular Modelling and Simulation”, Springer, 2010
- Additional:
- M.P. Allen and Tildesley, „Computer Simulation of Liquids“, Oxford Science Publishers. (Great book with a focus on Molecular Dynamics simulations)
- Frenkel und B. Smit. “Understanding molecular simulation” Academic, San Diego, 2002, (covers MD, MC and Stat Mech)
- Dill and S. Bromberg, “Molecular Driving Forces”, Taylor & Francis Inc, 2010
Software
www.gromacs.org open source molecular simulation software used in the tutorial, for both atomistic MD and coarse-grained Brownian dynamics simulations. Comes with an extensive manual, which includes the principles of MD simulations and biomolecular force fields.
www.poissonboltzmann.org open source software for continuum electrostatics calculations for biomolecules used in the tutorial on electrostatics.
sda7 open source software for Brownian dynamics simulation of the diffusional association of biomolecules using in the tutorial on Brownian dynamics.